Lignin
| Identifiers | |
|---|---|
CAS Number |
|
ChemSpider |
|
ECHA InfoCard | 100.029.699 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
Infobox references | |
Lignin is a class of complex organic polymers that form key structural materials in the support tissues of vascular plants and some algae.[1] Lignins are particularly important in the formation of cell walls, especially in wood and bark, because they lend rigidity and do not rot easily. Chemically, lignins are cross-linked phenolic polymers.[2]
Contents
1 History
2 Composition and structure
3 Biological function
4 Economic significance
5 Biosynthesis
6 Biodegradation
6.1 Lignin degradation by fungi
6.2 Lignin degradation by bacteria
7 Pyrolysis
8 Chemical analysis
9 References
10 External links
History
Lignin was first mentioned in 1813 by the Swiss botanist A. P. de Candolle, who described it as a fibrous, tasteless material, insoluble in water and alcohol but soluble in weak alkaline solutions, and which can be precipitated from solution using acid.[3] He named the substance “lignine”, which is derived from the Latin word lignum,[4] meaning wood. It is one of the most abundant organic polymers on Earth, exceeded only by cellulose. Lignin constitutes 30% of non-fossil organic carbon[5] and 20 to 35% of the dry mass of wood.[6] The Carboniferous Period (geology) is in part defined by the evolution of lignin.
Composition and structure
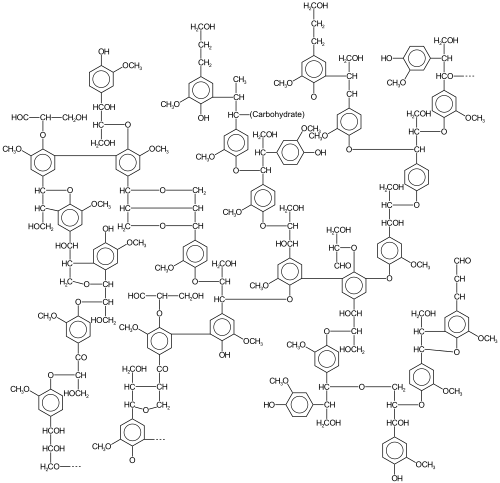
An example of a possible lignin structure. The portion shown here (correcting a C2H to CH2 and not counting the side chain denoted "Carbohydrate") has 28 monomers (mostly coniferyl alcohol), 278 carbon atoms, 407 hydrogen atoms, and 94 oxygen atoms (64% carbon, 8% hydrogen, and 29% oxygen by weight) which seems to be too high in hydrogen.
The composition of lignin varies from species to species. An example of composition from an aspen[7] sample is 63.4% carbon, 5.9% hydrogen, 0.7% ash (mineral components), and 30% oxygen (by difference),[8] corresponding approximately to the formula (C31H34O11)n.
As a biopolymer, lignin is unusual because of its heterogeneity and lack of a defined primary structure. Its most commonly noted function is the support through strengthening of wood (mainly composed of xylem cells and lignified sclerenchyma fibres) in vascular plants.[9][10][11]
The lignols that crosslink are of three main types, all derived from phenylpropane: 4-hydroxy-3-methoxyphenylpropane, 3,5-dimethoxy-4-hydroxyphenylpropane, and 4-hydroxyphenylpropane. The former tends to be more prevalent in conifers and the latter in hardwoods.
Lignin is a cross-linked polymer with molecular masses in excess of 10,000 u. It is relatively hydrophobic and rich in aromatic subunits. The degree of polymerisation is difficult to measure, since the material is heterogeneous. Different types of lignin have been described depending on the means of isolation.[12]

The three common monolignols: paracoumaryl alcohol (1), coniferyl alcohol (2) and sinapyl alcohol (3)
Three monolignol monomers are precursors, all of which are methoxylated to various degrees: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol[13]. These lignols are incorporated into lignin in the form of the phenylpropanoids p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S), respectively.[5]Gymnosperms have a lignin that consists almost entirely of G with small quantities of H. That of dicotyledonous angiosperms is more often than not a mixture of G and S (with very little H), and monocotyledonous lignin is a mixture of all three.[5] Many grasses have mostly G, while some palms have mainly S.[14] All lignins contain small amounts of incomplete or modified monolignols, and other monomers are prominent in non-woody plants.[15]
Biological function
Lignin fills the spaces in the cell wall between cellulose, hemicellulose, and pectin components, especially in vascular and support tissues: xylem tracheids, vessel elements and sclereid cells. It is covalently linked to hemicellulose and therefore cross-links different plant polysaccharides, conferring mechanical strength to the cell wall and by extension the plant as a whole.[16] It is particularly abundant in compression wood but scarce in tension wood, which are types of reaction wood.
Lignin plays a crucial part in conducting water in plant stems. The polysaccharide components of plant cell walls are highly hydrophilic and thus permeable to water, whereas lignin is more hydrophobic. The crosslinking of polysaccharides by lignin is an obstacle for water absorption to the cell wall. Thus, lignin makes it possible for the plant's vascular tissue to conduct water efficiently.[17] Lignin is present in all vascular plants, but not in bryophytes, supporting the idea that the original function of lignin was restricted to water transport. However, it is present in red algae, which seems to suggest that the common ancestor of plants and red algae also synthesised lignin. This would suggest that its original function was structural; it plays this role in the red alga Calliarthron, where it supports joints between calcified segments.[1]
Another possibility is that the lignins in red algae and in plants are a result of convergent evolution and not of a common origin.[1]
Economic significance

Pulp mill at Blankenstein, Germany. In such mills, using the kraft or the sulfite process, lignin is removed from lignocellulose to give pulp for papermaking.
Global commercial production of lignin is a consequence of papermaking. In 1988, more than 220 million tons of paper were produced worldwide.[18] Much of this paper was delignified; Lignin comprises about 1/3 of the mass of lignocellulose, the precursor to paper. It can thus be seen that lignin is handled on a very large scale. Lignin is an impediment to papermaking as it is colored, it yellows in air, and its presence weakens the paper. Once separated from the cellulose, it is burned as fuel. Only a fraction is used in a wide range of low volume applications where the form but not the quality is important.[19]
Mechanical, or high-yield pulp, which is used to make newsprint, contains most of the lignin originally present in the wood. This lignin is responsible for newsprint's yellowing with age.[4] High quality paper requires the removal of lignin from the pulp. These delignification processes are core technologies of the papermaking industry as well as the source of significant environmental concerns.
In sulfite pulping, lignin is removed from wood pulp as lignosulfonates, for which many applications have been proposed.[20] They are used as dispersants, humectants, emulsion stabilizers, and sequestrants (water treatment).[21]
Lignin removed by the kraft process is usually burned for its fuel value, providing energy to power the mill. Two commercial processes exist to remove lignin from black liquor for higher value uses: LignoBoost (Sweden) and LignoForce (Canada). Higher quality lignin presents the potential to become a renewable source of aromatic compounds for the chemical industry, with an addressable market of more than $130bn.[22]
Given that it is the most prevalent biopolymer after cellulose, lignin has been investigated as a feedstock for biofuel production.[23][24]
Biosynthesis
Lignin biosynthesis begins in the cytosol with the synthesis of glycosylated monolignols from the amino acid phenylalanine. These first reactions are shared with the phenylpropanoid pathway. The attached glucose renders them water-soluble and less toxic. Once transported through the cell membrane to the apoplast, the glucose is removed, and the polymerisation commences.[25] Much about its anabolism is not understood even after more than a century of study.[5]

Polymerisation of coniferyl alcohol to lignin. The reaction has two alternative routes catalysed by two different oxidative enzymes, peroxidases or oxidases.
The polymerisation step, that is a radical-radical coupling, is catalysed by oxidative enzymes. Both peroxidase and laccase enzymes are present in the plant cell walls, and it is not known whether one or both of these groups participates in the polymerisation. Low molecular weight oxidants might also be involved. The oxidative enzyme catalyses the formation of monolignol radicals. These radicals are often said to undergo uncatalyzed coupling to form the lignin polymer, but this hypothesis has been recently challenged.[26] The alternative theory that involves an unspecified biological control is however not widely accepted.
Biodegradation
In contrast to other bio-polymers (e.g. proteins, DNA, and even cellulose), lignin is resistant to degradation and acid- and base-catalyzed hydrolysis. However the extent to which lignin does or does not degrade varies with species and plant tissue type. For example, syringyl (S) lignol is more susceptible to degradation by fungal decay as it has fewer aryl-aryl bonds and a lower redox potential than guaiacyl units.[27][28] Because it is cross-linked with the other cell wall components, lignin minimizes the accessibility of cellulose and hemicellulose to microbial enzymes, leading to a reduced digestibility of biomass.[17]
Some ligninolytic enzymes include heme peroxidases such as lignin peroxidases, manganese peroxidases, versatile peroxidases, and dye-decolourizing peroxidases as well as copper-based laccases. Lignin peroxidases oxidize non-phenolic lignin, whereas manganese peroxidases only oxidize the phenolic structures. Dye-decolorizing peroxidases, or DyPs, exhibit catalytic activity on a wide range of lignin model compounds, but their in vivo substrate is unknown. In general, laccases oxidize phenolic substrates but some fungal laccases have been shown to oxidize non-phenolic substrates in the presence of synthetic redox mediators.[29][30]
Lignin degradation by fungi
Well-studied ligninolytic enzymes are found in Phanerochaete chrysosporium[31] and other white rot fungi. Some white rot fungi, such as C. subvermispora, can degrade the lignin in lignocellulose, but others lack this ability. Most fungal lignin degradation involves secreted peroxidases. Many fungal laccases are also secreted, which facilitate degradation of phenolic lignin-derived compounds, although several intracellular fungal laccases have also been described. An important aspect of fungal lignin degradation is the activity of accessory enzymes to produce the H2O2 required for the function of lignin peroxidase and other heme peroxidases.[29]
Lignin degradation by bacteria
Bacteria lack most of the enzymes employed in fungal lignin degradation, yet bacterial degradation can be quite extensive.[32] The ligninolytic activity of bacteria has not been studied extensively even though it was first described in 1930. Many bacterial DyPs have been characterized. Bacteria do not express any of the plant-type peroxidases (lignin peroxidase, Mn peroxidase, or versatile peroxidases), but three of the four classes of DyP are only found in bacteria. In contrast to fungi, most bacterial enzymes involved in lignin degradation are intracellular, including two classes of DyP and most bacterial laccases.[30]
Bacterial degradation of lignin is particularly relevant in aquatic systems such as lakes, rivers, and streams, where inputs of terrestrial material (e.g. leaf litter) can enter waterways and leach dissolved organic carbon rich in lignin, cellulose, and hemicellulose. In the environment, lignin can be degraded either biotically via bacteria or abiotically via photochemical alteration, and oftentimes the latter assists in the former.[33] In addition to the presence or absence of light, several of environmental factors affect the biodegradability of lignin, including bacterial community composition, mineral associations, and redox state.[34][35]
Pyrolysis
Pyrolysis of lignin during the combustion of wood or charcoal production yields a range of products, of which the most characteristic ones are methoxy-substituted phenols. Of those, the most important are guaiacol and syringol and their derivatives. Their presence can be used to trace a smoke source to a wood fire. In cooking, lignin in the form of hardwood is an important source of these two compounds, which impart the characteristic aroma and taste to smoked foods such as barbecue. The main flavor compounds of smoked ham are guaiacol, and its 4-, 5-, and 6-methyl derivatives as well as 2,6-dimethylphenol. These compounds are produced by thermal breakdown of lignin in the wood used in the smokehouse.[36]
Chemical analysis
The conventional method for lignin quantitation in the pulp industry is the Klason lignin and acid-soluble lignin test, which is standardized according to TAPPI[37] or NREL[38] procedure. The cellulose is first decrystallized and partially depolymerized into oligomers by keeping the sample in 72% sulfuric acid at 30 C for 1 h. Then, the acid is diluted to 4% by adding water, and the depolymerization is completed by either boiling (100 °C) for 4 h or pressure cooking at 2 bar (124 °C) for 1 h. The acid is washed out and the sample dried. The residue that remains is termed Klason lignin. A part of the lignin, acid-soluble lignin (ASL) dissolves in the acid. ASL is quantified by the intensity of its UV absorption peak at 280 nm. The method is suited for wood lignins, but not equally well for varied lignins from different sources. The carbohydrate composition may be also analyzed from the Klason liquors, although there may be sugar breakdown products (furfural and 5-hydroxymethylfurfural).
A solution of hydrochloric acid and phloroglucinol is used for the detection of lignin (Wiesner test). A brilliant red color develops, owing to the presence of coniferaldehyde groups in the lignin.[39]
Thioglycolysis is an analytical technique for lignin quantitation.[40] Lignin structure can also be studied by computational simulation.[41]
Thermochemolysis (chemical break down of a substance under vacuum and at high temperature) with tetramethylammonium hydroxide (TMAH) or cupric oxide[42] has also been used to characterize lignin chemical composition. The ratio of syringyl lignol (S) to vanillyl lignol (V) and cinnamyl lignol (C) to vanillyl lignol (V) is variable based on plant type and can therefore be used to trace plant sources in aquatic systems (woody vs. non-woody and angiosperm vs. gymnosperm).[43] Ratios of carboxylic acid (Ad) to aldehyde (Al) forms of the lignols (Ad/Al) reveal diagenetic information, with higher ratios indicating a more highly degraded material.[44][45] Increases in the (Ad/Al) value indicate an oxidative cleavage reaction has occurred on the alkyl lignin side chain which has been shown to be a step in the decay of wood by many white-rot and some soft rot fungi.[44][45][46][47][48]
Solid state 13C NMR has been used to look at the concentrations of lignin, as well as other major components in wood e.g. cellulose, and how that changes with microbial decay.[44][45][47][48] Conventional solution-state NMR for lignin is possible. However, many intact lignins have a crosslinked, very high molar-mass fraction that is difficult to dissolve even for functionalization.
References
^ abc Martone, Pt; Estevez, Jm; Lu, F; Ruel, K; Denny, Mw; Somerville, C; Ralph, J (Jan 2009). "Discovery of Lignin in Seaweed Reveals Convergent Evolution of Cell-Wall Architecture". Current Biology. 19 (2): 169–75. doi:10.1016/j.cub.2008.12.031. ISSN 0960-9822. PMID 19167225..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Lebo, Stuart E. Jr.; Gargulak, Jerry D.; McNally, Timothy J. (2001). "Lignin". Kirk-Othmer Encyclopedia of Chemical Technology. Kirk‑Othmer Encyclopedia of Chemical Technology. John Wiley & Sons, Inc. doi:10.1002/0471238961.12090714120914.a01.pub2. ISBN 978-0-471-23896-6. Retrieved 2007-10-14.
^ de Candolle, M.A.P. (1813). Theorie Elementaire de la Botanique ou Exposition des Principes de la Classification Naturelle et de l'Art de Decrire et d'Etudier les Vegetaux. Paris: Deterville.
^ ab E. Sjöström (1993). Wood Chemistry: Fundamentals and Applications. Academic Press. ISBN 978-0-12-647480-0.
^ abcd W. Boerjan; J. Ralph; M. Baucher (June 2003). "Lignin biosynthesis". Annu. Rev. Plant Biol. 54 (1): 519–549. doi:10.1146/annurev.arplant.54.031902.134938. PMID 14503002.
^ Li Jingjing (2011) Isolation of Lignin from Wood. SAIMAA UNIVERSITY OF APPLIED SCIENCES.
^ In the referenced article, the species of aspen is not specified, only that it was from Canada.
^ Hsiang-Hui King; Peter R. Solomon; Eitan Avni; Robert W. Coughlin (Fall 1983). "Modeling Tar Composition in Lignin Pyrolysis" (PDF). Symposium on Mathematical Modeling of Biomass Pyrolysis Phenomena, Washington, D.C., 1983. p. 1.
^ (1995, Biology, Arms and Camp ).
^ Anatomy of Seed Plants, Esau, 1977
^ Wardrop; The (1969). "Eryngium sp.;". Aust. J. Botany. 17 (2): 229–240. doi:10.1071/bt9690229.
^ "Lignin and its Properties: Glossary of Lignin Nomenclature". Dialogue/Newsletters Volume 9, Number 1. Lignin Institute. July 2001. Retrieved 2007-10-14.
^ K. Freudenberg & A.C. Nash (eds) (1968). Constitution and Biosynthesis of Lignin. Berlin: Springer-Verlag.CS1 maint: Extra text: authors list (link)
^ Kuroda K, Ozawa T, Ueno T (April 2001). "Characterization of sago palm (Metroxylon sagu) lignin by analytical pyrolysis". J Agric Food Chem. 49 (4): 1840–7. doi:10.1021/jf001126i. PMID 11308334.
^ J. Ralph; et al. (2001). "Elucidation of new structures in lignins of CAD- and COMT-deficient plants by NMR". Phytochemistry. 57 (6): 993–1003. doi:10.1016/S0031-9422(01)00109-1. PMID 11423146.
^ Chabannes, M.; et al. (2001). "In situ analysis of lignins in transgenic tobacco reveals a differential impact of individual transformations on the spatial patterns of lignin deposition at the cellular and subcellular levels". Plant J. 28 (3): 271–282. doi:10.1046/j.1365-313X.2001.01159.x. PMID 11722770.
^ ab K.V. Sarkanen & C.H. Ludwig (eds) (1971). Lignins: Occurrence, Formation, Structure, and Reactions. New York: Wiley Intersci.CS1 maint: Extra text: authors list (link)
^ Rudolf Patt et al. (2005). "Pulp". Paper and Pulp. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. pp. 1–92. doi:10.1002/14356007.a18_545.pub4. ISBN 9783527306732.
^ NNFCC Renewable Chemicals Factsheet: Lignin
^ "Uses of lignin from sulfite pulping". Archived from the original on 2007-10-09. Retrieved 2007-09-10.
^ Barbara A. Tokay (2000). "Biomass Chemicals". Ullmann's Encyclopedia Of Industrial Chemistry. doi:10.1002/14356007.a04_099. ISBN 978-3527306732.
^ "Frost & Sullivan: Full Speed Ahead for the Lignin Market with High-Value Opportunities as early as 2017".
^ Folkedahl, Bruce (2016), "Cellulosic ethanol: what to do with the lignin", Biomass, retrieved 2016-08-10.
^ Abengoa (2016-04-21), The importance of lignin for ethanol production, retrieved 2016-08-10.
^ Samuels AL, Rensing KH, Douglas CJ, Mansfield SD, Dharmawardhana DP, Ellis BE (November 2002). "Cellular machinery of wood production: differentiation of secondary xylem in Pinus contorta var. latifolia". Planta. 216 (1): 72–82. doi:10.1007/s00425-002-0884-4. PMID 12430016.
^ Davin, L.B.; Lewis, N.G. (2005). "Lignin primary structures and dirigent sites". Current Opinion in Biotechnology. 16 (4): 407–415. doi:10.1016/j.copbio.2005.06.011. PMID 16023847.
^ Vane, Christopher H.; Drage, Trevor C.; Snape, Colin E. (February 2003). "Biodegradation of Oak Wood during Growth of the Shiitake Mushroom: A Molecular Approach". Journal of Agricultural and Food Chemistry. 51 (4): 947–956. doi:10.1021/jf020932h. PMID 12568554.
^ Vane, Christopher H.; Drage, Trevor C.; Snape, Colin E. (January 2006). "Bark decay by the white-rot fungus Lentinula edodes: Polysaccharide loss, lignin resistance and the unmasking of suberin". International Biodeterioration & Biodegradation. 57 (1): 14–23. doi:10.1016/j.ibiod.2005.10.004.
^ ab Advances in applied microbiology. Vol. 82. Gadd, Geoffrey M., Sariaslani, Sima. Oxford: Academic. 2013. pp. 1–28. ISBN 9780124076792. OCLC 841913543.
^ ab de Gonzalo, Gonzalo; Colpa, Dana I.; Habib, Mohamed H.M.; Fraaije, Marco W. (2016). "Bacterial enzymes involved in lignin degradation". Journal of Biotechnology. 236: 110–119. doi:10.1016/j.jbiotec.2016.08.011. PMID 27544286.
^ Tien, M (1983). "Lignin-Degrading Enzyme from the Hymenomycete Phanerochaete chrysosporium Burds". Science. 221 (4611): 661–3. doi:10.1126/science.221.4611.661. PMID 17787736.
^ Pellerin, Brian A.; Hernes, Peter J.; Saraceno, JohnFranco; Spencer, Robert G. M.; Bergamaschi, Brian A. (May 2010). "Microbial degradation of plant leachate alters lignin phenols and trihalomethane precursors". Journal of Environmental Quality. 39 (3): 946–954. doi:10.2134/jeq2009.0487. ISSN 0047-2425. PMID 20400590.
^ Hernes, Peter J. (2003). "(PDF) Photochemical and microbial degradation of dissolved lignin phenols: Implications for the fate of terrigenous dissolved organic matter in marine environments". Journal of Geophysical Research. 108. doi:10.1029/2002JC001421. Retrieved 2018-11-27.
^ "(PDF) Persistence of Soil Organic Matter as an Ecosystem Property". ResearchGate. Retrieved 2018-11-27.
^ Dittmar, Thorsten (2015-01-01). Reasons Behind the Long-Term Stability of Dissolved Organic Matter. Biogeochemistry of Marine Dissolved Organic Matter. pp. 369–388. doi:10.1016/B978-0-12-405940-5.00007-8. ISBN 9780124059405.
^ Wittkowski, Reiner; Ruther, Joachim; Drinda, Heike; Rafiei-Taghanaki, Foroozan "Formation of smoke flavor compounds by thermal lignin degradation" ACS Symposium Series (Flavor Precursors), 1992, volume 490, pp 232–243.
ISBN 978-0-8412-1346-3.
^ TAPPI. T 222 om-02 - Acid-insoluble lignin in wood and pulp
^ Sluiter, A., Hames, B., Ruiz, R., Scarlata, C., Sluiter, J., Templeton, D., Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass. Technical Report NREL/TP-510-42618, U.S. Department of Energy, 2008. [1]
^ Lignin production and detection in wood. John M. Harkin, U.S. Forest Service Research Note FPL-0148, November 1966 (article)
^ Lange, B. M.; Lapierre, C.; Sandermann, Jr (1995). "Elicitor-Induced Spruce Stress Lignin (Structural Similarity to Early Developmental Lignins)". Plant Physiology. 108 (3): 1277–1287. doi:10.1104/pp.108.3.1277. PMC 157483. PMID 12228544.
^ Glasser, Wolfgang G.; Glasser, Heidemarie R. (1974). "Simulation of Reactions with Lignin by Computer (Simrel). II. A Model for Softwood Lignin". Holzforschung. 28 (1): 5–11, 1974. doi:10.1515/hfsg.1974.28.1.5.
^ Hedges, John I.; Ertel, John R. (February 1982). "Characterization of lignin by gas capillary chromatography of cupric oxide oxidation products". Analytical Chemistry. 54 (2): 174–178. doi:10.1021/ac00239a007. ISSN 0003-2700.
^ Hedges, John I.; Mann, Dale C. (1979-11-01). "The characterization of plant tissues by their lignin oxidation products". Geochimica et Cosmochimica Acta. 43 (11): 1803–1807. doi:10.1016/0016-7037(79)90028-0. ISSN 0016-7037.
^ abc Vane, C. H.; et al. (2003). "Biodegradation of Oak (Quercus alba) Wood during Growth of the Shiitake Mushroom (Lentinula edodes): A Molecular Approach". Journal of Agricultural and Food Chemistry. 51 (4): 947–956. doi:10.1021/jf020932h. PMID 12568554.
^ abc Vane, C. H.; et al. (2006). "Bark decay by the white-rot fungus Lentinula edodes: Polysaccharide loss, lignin resistance and the unmasking of suberin". International Biodeterioration & Biodegradation. 57 (1): 14–23. doi:10.1016/j.ibiod.2005.10.004.
^ Vane, C. H.; et al. (2001). "The effect of fungal decay (Agaricus bisporus) on wheat straw lignin using pyrolysis–GC–MS in the presence of tetramethylammonium hydroxide (TMAH)". Journal of Analytical and Applied Pyrolysis. 60 (1): 69–78. doi:10.1016/s0165-2370(00)00156-x.
^ ab Vane, C. H.; et al. (2001). "Degradation of Lignin in Wheat Straw during Growth of the Oyster Mushroom (Pleurotus ostreatus) Using Off-line Thermochemolysis with Tetramethylammonium Hydroxide and Solid-State 13C NMR". Journal of Agricultural and Food Chemistry. 49 (6): 2709–2716. doi:10.1021/jf001409a.
^ ab Vane, C. H.; et al. (2005). "Decay of cultivated apricot wood (Prunus armeniaca) by the ascomycete Hypocrea sulphurea, using solid state 13C NMR and off-line TMAH thermochemolysis with GC–MS". International Biodeterioration & Biodegradation. 55 (3): 175–185. doi:10.1016/j.ibiod.2004.11.004.
External links
- Tecnaro website
Paper | |
|---|---|
| |
| Types |
|
| Materials |
|
| Specifications |
|
| Manufacture and process |
|
| Industry |
|
| |
Wood products | |
|---|---|
Lumber/ timber |
|
| Engineered wood |
|
| Fuelwood |
|
| Fibers |
|
| Derivatives |
|
| By-products |
|
| Historical |
|
| See also |
|
| |
Authority control |
|
|---|


